有关DTA的基本概念/相关基础知识下面不做阐述。下面做介绍的是我觉得比较重要/有趣的内容。
第一部分:DTA的发展
(1)高通量筛选(High-Throughput Screening, HTS)
核心逻辑:在生物或生化实验中,利用自动化设备对数以万计的化学化合物进行大规模的靶点相互作用测试。
当前阶段痛点:实验成本极其高昂且耗时。目前存在数以百万计的可用药物化合物和数千个潜在靶点蛋白,传统的 HTS 根本无法覆盖如此庞大的配对搜索空间。
(2)分子对接(molecular docking)
核心逻辑:基于受体(蛋白质靶点)和配体(药物分子)精确的三维 (3D) 晶体结构,计算其结合姿态 (Binding Pose),并使用物理或经验打分函数来预测结合亲和力。
当前阶段痛点:严重依赖蛋白质和化合物精确的 3D 结构。然而,获取许多靶点蛋白的高分辨率 3D 结构非常困难。此外,对接过程对结构极度敏感,微小的构象变化会导致结果产生巨大差异,因此不仅计算开销大,且不适合探索具有相似结构的未知靶点空间。
(3)传统机器学习
核心逻辑:假设药物与靶点之间的亲和力可以通过人工设计的特征描述符(Descriptors)来预测。通过提取药物的分子指纹和蛋白质的理化/序列性质,送入随机森林 (RF)、支持向量机 (SVM) 或多重线性回归 (MLR) 等模型预测打分。
当前阶段痛点:无法自动提取高阶的隐藏特征。模型性能严重依赖于领域专家手工构建的特征(Feature Engineering),在这个中间降维过程中容易丢失关键的分子拓扑或空间信息。同时,一些模型仅基于药物特征构建,只能探索药物空间,而无法泛化到未知的靶点空间。
(4)深度学习
核心逻辑:将特征表征提取和目标值预测无缝集成在同一个架构中。可以直接将药物的 SMILES 字符串或分子图,以及蛋白质的氨基酸序列或接触图(Contact Map)输入到 CNN、RNN、GCN 或 Transformer 等网络中,模型会自动学习深层表征。
当前阶段的痛点:可解释性差、泛化能力差等(列举的这两个痛点应该是现在DL领域普遍面临的问题)
第二部分:DTA领域遇到的挑战
下面结论据不完全统计。
(1)泛化能力不够强
很多模型在常见 benchmark 上分数很高,但一旦换成新靶点、新骨架、低相似度样本,或者采用更严格的数据切分,性能就会明显下降。
(2)模型离真实结合物理过程还较远
DTA 不是简单的“药物图 + 蛋白序列 -> 一个数”。真实结合过程涉及蛋白柔性、诱导契合、口袋构象变化、溶剂效应、熵、甚至结合/解离动力学。但很多现有方法仍然主要依赖静态序列或单一结构快照,因此即使拟合了数据,也未必真的学到了可迁移的结合机制。这也是为什么“从静态表征走向动态、机制驱动建模”被认为是长期瓶颈。
(3)数据与标签本身不够可靠
DTA 的标签往往来自不同实验体系和条件,像IC50、Ki、Kd之间并不完全等价;不同assay、批次、实验噪声、负样本构造方式,都会把“真实亲和力”污染成一个很嘈杂的监督信号。再加上高质量数据总体仍然稀缺、覆盖不均衡,模型很容易学到偏差而不是规律。因此,数据稀缺、标签噪声、异质性和不确定性校准,是第三个顶层问题。
第三部分:用于DTA的DL模型面临的部分更细化的挑战
Ps:我这边只列于了后面要用到的一些痛点,也是这些后续工作的重点攻克的问题。
(1)深层交互丢失
“Current DTI prediction methods typically develop a single fusion module, which often integrates features through concatenation or weighting. This strategy struggles to capture deep interactions… and easily loses critical interaction information.”
cite:Cao Z, Xie J, Xu J, Li B. Cross-Modal Interaction-Aware Progressive Fusion Network for Drug-Target Interaction Prediction. J Chem Inf Model. 2025 Oct 13;65(19):10713-10726. doi: 10.1021/acs.jcim.5c01429. Epub 2025 Sep 19. PMID: 40970765.
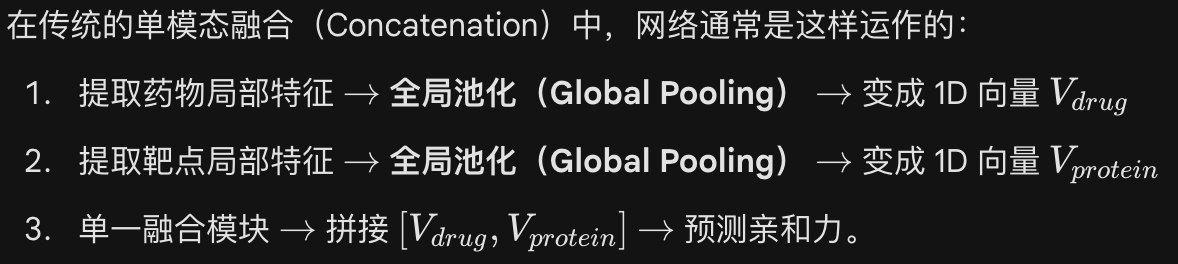
现有的 DTA 预测模型通常采用后期特征拼接(Late Concatenation)作为单一的融合策略。这个痛点关键在于之前的DTA模型的“全局池化”发生在了“特征融合”之前。当你把拥有 3D 空间结构的蛋白质压缩成一个 1D 向量时,里面具体的“口袋凹槽”、“氢键供体/受体位置”就被抹平了。此时再去和药物拼接,模型根本无法建立“药物的羟基到底是对准了靶点的哪一个氨基酸”这种深层空间对应关系。这就是文献所说的 “loses critical interaction information”。
(2)弱监督下的宏观定位与背景噪声干扰(信号稀释)
*“Predicting drug-target interactions (DTI) with graph neural networks (GNNs) is hindered by their lack of interpretability… Nevertheless, adoption of GNNs in drug discovery remains limited due to their ‘black-box’ nature and the inability to provide chemically intuitive explanations for predictions.”
cite:Mahindran, M., Liu, Q., Kadambalithaya, V. M., & Kalinina, O. V. (2026). Explainability methods from machine learning detect important drugs’ atoms in drug-target interactions[Preprint]. bioRxiv
这个问题其实就是上面的标签噪声问题。
深层原因(数据局限):实验数据往往只能提供全局的亲和力标签,缺乏原级/残基级别的微观结合位点标注(即弱监督困境)。传统的网络架构在面对这种数据时,只能进行粗暴的“全局特征拼接 -> 全局标签预测”,彻底丢失了特征溯源的能力。
这个“black-box”会导致一个问题,就是我们在拿到模型的高分预测后,依然不知道该对分子的哪个基团进行修饰,对后面从苗头化合物向先导化合物产生困难。(“知其然不知其所以然”)